NEWSLETTER OF eurostar-science | |
NO 4 -- September 29, 2002 |
CONTENTS
4.1 EDITORIAL| 4.1 EDITORIAL Erwin Marti, President |
||||
|
The
PhandTA
6
is
over,
let
us
plan
the
next
PhandTA. Erwin Marti, President |
||||
| 4.2
Highlights
of
the
PhandTA
6 Monte Veritŕ, Ascona, Switzerland Conference: May 26-29, 2002 Workshops: May 30, 2002 |
||||
|
The
PhandTA
6
was
the
second
Conference
on
the
subject
Pharmacy
and
Applied
Physical
Chemistry
which
was
held
at
the
Conference
Center
"Stefano
Franscini"
at
Monte
Veritŕ
above
Ascona.
The
anticipated
Scientific
Program
was
arranged
according
to
the
following
sections: About 125 Participants from 22 Countries attended the Conference. The 72 Communications consisted of 50 Lectures including the Communications presented during the Workshops and additional 22 Poster Presentations.Two Awards have been dedicated at the Conference. The Award for "Applied Physical Chemistry" of eurostar-science for the year 2002 has been dedicated to Professor Stephan Grzesiek, Department of Structural Biology, Biocenter of the University of Basel, for his research in the area of the elucidation of biological macromolecules using methods of Nuclear Magnetic Resonance. The classical pathway in the elucidation of protein structure was to try to crystallize the proteins and to study the solid state structure in the way they diffracted x-rays. Stephan Grzesiek`s great innovation was to try to study the interactions and the structure of proteins in solution with 13C atoms of the amino acids side chains and the 1H and 15N atoms of the protein main chain. The break-through in this structure elucidation was demonstrated with interferon-g, having a molecular weight of 31'400. Another highlight coming out as a result from the laboratory of Stephan Grzesiek is the attempt to measure, for the first time, the distances of hydrogen bonds in nucleic acids and proteins in solution. The Award for "Applied Chemical Thermodynamics" of the Swiss Society for Thermal Analysis and Calorimetry has been presented for the year 2001 at the PhandTA 6 to Dr. Erwin Marti, Basel, for his project work in the chemical and pharmaceutical industry in several different areas of the physical chemistry. Highlights of his work are based on the determination of thermodynamic data and their evaluation such as partial and vapor pressure, melting temperature, heat of fusion and sublimation, molar heat capacities, phase diagrams, purity methods (DSC and Phase solubility), solubility and dissolution rate, chemical reactions and reaction pathways, physical instabilities caused by polymorphic changes and re-crystallization of crystalline powders. In 1973, the absolute stability was introduced for polymorphs of organic substances with the Gibb`s free energy. These functions overcome handicaps and troubles of mere kinetic observations because they are on the base of equilibrium results. All these activities lead often to the introduction or development of a new method, to a new analytical evaluation procedure, to a better chemical or physical production pathway for active substances from different domains. Some of the initiated projects lead to patents for new chemical entities in areas with rather important chemical and therapeutic applications. Professor Peter York, University of Bradford, UK was presenting the Opening Lecture as a Review about "Particle Design for Drug Delivery Systems" with the aim to produce drug substances as crystals with a given corn size and corn size distribution. Low solubility values of a given drug or as an example administration in the respiratory track afford fine or even ultra-fine particles in the region of microns or even below. The classical production pathway with a targeted fine particle size is a multistage process with a crystallization or precipitation followed by harvesting of the finest achieved crystals, drying, consecutive milling or micronizing. Often, to much energy is transferred to the crystals by such a milling process and locally high temperature spots could cause a chemical decomposition. In addition, an amorphisation and also a polymorphic transformation could be initiated by milling. Supercritical fluid methods in a combination with an anti-solvent used twofold, namely as dispersing phase for the drug substance and as extracting phase for the solvent has been developed in Peter York`s group. Such a process can yield to considerable quantities of drug substances with a high quality in several aspects (purity and low solvent content) and after an optimization fine crystal powders are achieved with a narrow size distribution and a selected median value within a range of 0.5 to 30 microns. Professor Dario Anselmetti, University of Bielefeld, Germany gave a lecture on "Biophysical Analysis of Single Molecules". The main interest of his group is directed towards Atomic Force Microscopy (AFM) and bioanalytical micro- and nano-fluidics, the latter being also a highly sophisticated research area. He explained as introduction the concepts of detecting, addressing and manipulating of individual molecules by mechanical experiments (AFM), by optical fluorescence microscopy, and by interactions with electrical fields. Several examples were outlined among others a molecular switch using an ionic interaction of Fe3+ with the biomolecule under investigation, and the influence on the force histogram by an interaction of Galacto- and Succinoglucan respectively with a DNA molecule. Dr. Rolf Hilfiker, Solvias AG, Basel, CH has outlined in a Plenary Lecture with the title "High Throughput Polymorph Screening (HTS): Just the First Step for Integrated Crystal Engineering" the position of this method within the elucidation of co-crystals and polymorphs for a chemical entity under investigation. To know the existence of the most important co-crystal and the polymorphs of a substance is a crucial point and as an example for a drug substance properties such as bioavailability, processability, the physical and chemical stability may reveal marked differences. The concept of the HTS is the crystallization of an active substance e.g. a drug substance from a solution or from a suspension by controlled evaporation of the solvent for up to 96 samples on a micro-titer plate made from quartz. The miniaturized experiments afford a rather small amount of the active substance, the experimental procedure is straightforward and so is the characterization with Raman microscopy as the method of choice. Additional characterizations such as hot-stage Raman Microscopy and thermomicroscopy enable an additional information of the obtained substances and polymorphs. The High Throughput Screening must be regarded as an first step in an elucidation of the solid state variety of a drug substance. The screening reveals the potential of the varieties for a given substance, however, depending on the goals and needs of research and development, well selected further investigations have to follow. Professor Joachim Seelig, Biocenter of the University of Basel reported on "The Lipid Bilayer. Specific and Non-specific Interactions with Surfactants, Peptides and Proteins". The structure and the functions of the biological membranes has been outlined and also their importance for any living cell and any intracellular compartments. Membranes are based on a lipid bilayer sheath with a thickness of about 10 to 15 nm. Proteins are embedded in the lipid bilayer creating properties of smectic liquid crystal. These membranes have complex functions: Transport of hydrophobic and hydrophilic substances, of ions. Such transports are often against a concentration gradient (active transport). Membranes are also existing with a strict barrier for molecular sizes above a certain limit (blood-brain barrier). The structure of membranes are preferably elucidated by electron microscopy and by solid-state nuclear magnetic resonance.Interactions of membranes with surfactants or with peptides are nowadays studied with high sensitivity calorimeters yielding a complete set of thermodynamic data for the complex formed: The enthalpy of binding, the binding constant K and the free energy of binding ∆G, the entropy of binding ∆S, the stoichiometric ratio n. Most of the interactions are non-specific and can be described by membrane-liquid phase partition equilibrium or a partition coefficient. The analysis requires in addition the consideration of electrostatic effects (electrical double layer: Helmholtz, Gouy-Chapman, Stern). Examples were given for the interaction with surfactants, for an antimicrobal peptide which forms helices in contact with the membrane surface, and also for Alzheimer peptides. The latter undergo a membrane-induced random coil-β-structure transition. The antimicrobial peptide cinnamycin is an example for a specific interaction with a particular lipid, phosphatidylethanolamine. Dr. Katharina Maria Picker, Martin-Luther-University Halle-Wittenberg, Germany gave a Plenary Lecture about the subject "Mechanical and Physico-Chemical Characterization of Excipients in Order to Define their Applicability for Tabletting" as well as a communication about tabletting at a Workshop. The cardinal goals of the process of tablet production always was to formulate tablets which are robust, as stable as possible and also administered with a high repro-ducibility. To achieve these goals it was necessary to add excipients which could embody the crystalline drug substances during the deformation process. The drug substance is, of course, during the tabletting process also exposed to mechanical forces as well as to an increased temperature inside the formed tablet. This temperature rise depends on the tablet size and composition, on the tabletting machine used and their working adjustments. The temperature rise can be considerably high for certain small volume fractions within each tablet. Of course, these temperature rise in small domains is strictly time limited over the period of the tabletting process (increasing density of the tablet formed) and over a short additional time interval after the release of the tablet from the press. There are simply spoken two classes of drug substances, the first class rather insensitive to tabletting and the second mainly also enzymes, proteins and certain low molecular drugs which afford a so-called "soft tabletting". Changes of the drug substances which could occur during this process are chemical decomposition, polymorphic changes, formation or increasing of the amorphous content. These rather complex processes were studied with selected systems using different classes of excipients (microcrystalline cellulose, dicalcium phosphate dihydrate, hydroxypropyl methyl-cellulose, carrageenans, chitosans, polyethylene oxides) and an eccentric tabletting machine was used. The results reveal the fact that the mechanical properties of the used excipients have a great impact on the quality of a given drug substance in the final tablet and there durability. Professor Michael J. Pikal, University of Connecticut, Storrs, USA introduced into "Freeze Drying of Pharmaceuticals: A Review of Critical Parameters" and he gave also a survey at a Workshop on development work performed in his laboratory on freeze drying. Drug substances which are not extremely robust as biomolecules with instability in solution, at elevated temperature or difficulty in their crystallization are candidates for freeze drying. The production process starts always with a solution of the drug substances normally in water, however, also non aqueous solvents are used with one or several excipients. These components are protective colloids such as dextran, PVP and sugars such as sucrose, maltose, lactose, stabilizers as sodium glutamate, puffers and electrolytes. The production pathway is rather complex starting with cooling of a vial with the appropriate formulation, supercooling, nucleation and crystallization of water with a concentration of the solutes. A crystallization of solutes and especially of the drug substance may occur at a final stage of freeze drying or also a formation of amorphous material. Crystallization of the amorphous phases could be also possible during the annealing interval. There are crucial points during theses processes, some interrelated with each other as the rate of the water evaporation determined among other factors by the temperature of the surface of the solution in the vial (vapor pressure of water in the solution), the absolute shelf temperature during freezing. Process control is critical to minimize the process time while maintaining product quality. The collapse temperature , for which the temperature of the freeze dried material becomes identical with its glass transition temperature (depending also on the water content) should be avoided by the process pathway or, if necessary by an appropriate formulation.The stability of the freeze dried product is not easy to understand, because of the different physical phases and the possible influence of all the components. The main concept of freeze drying is to try to disperse a drug substance in a amorphous matrix with a rather low water content. These matrixes form a structurally rather rigid environment and also a non reactive phase. These matrixes are revealing a physical relaxation process during storage, which can be investigated with isothermal calorimetry. These kinetic processes are depending among other factors on the composition and nature of the amorphous phase, storage temperature, moisture content. ( see also the Report of the sponsor presented in this Newsletter, namely TA instruments, by R. Bruce Cassel on physical Aging of amorphous Sucrose). The Plenary Lecture of Professor Hans-Beat Bürgi, University of Bern, CH covered with "Physicochemical Aspects of Polymorph Prediction" an extremely interesting subject. Several groups of theoretical crystallographers, mainly from the Academia, made extraordinarily attempts to predict all the stable polymorphs of organic substances without incorporation of any experimental data, starting with the structure of the molecule alone. As a conclusion of these basic research work Professor Bürgi made the statement: None of these theoretical schemes has succeeded in consistently identifying from such sets the polymorphs of an organic substance appearing on crystallization. On the other hand, to elaborate computer codes for generating sets of energetically and geometrically reasonable crystal structures have proven very helpful in characterizing materials for which diffraction information is limited e.g. for poorly crystalline materials with available powder data. Pathways were discussed to improve the theoretical approaches, one being the attempts to understand the crystallization processes including nucleation, growth mechanisms and disordering phenomena. These latter processes are extremely complex with formation of the nuclei and their equilibrium state, their possible phase transition in this transient states. These steps are of greatest influence for the formation of the polymorph of the final crystal. All of these topics are ideal themes for fundamental research. Examples of handicaps of the present theoretical approaches in predicting polymorphs have been outlined. Dr. Samuel Petit, University of Rouen, F presented the subject "Oscillating Crystallization of a Chiral Compound in Quasi-racemic Solution; Evidence of a Multiepitaxy Crystal Growth Mechanism and Influence by Stirring" and he outlined additionally more general aspects on crystal growth at a Workshop. Preferential crystallization is an efficient process for the preparative separation of chiral drug substances or intermediates. The implementation of such a process requires that the racemic mixture crystallizes as a conglomerate or in other words in a mechanical mixture of crystals containing only one enantiomer. A study of the binary phase diagram of the enantiomers may reveal the existence of an eutectic system which is a prerequisite for a separation (see also the poster presentation by Judith Rollinger, Ulrich Griesser, Martin Szelagiewicz, and Urs Hofmeier on "Thermoanalytical Investigation of the complex Binary System of (+)- and (-)- Diprophylline").An example was presented with an eutectic phase diagram, however, the separation in water as liquid phase was of a rather poor yield. A crystal growth study , combined with stereospecific dissolution experiments revealed that each crystal was made of homochiral lamellar zones. Therefore, the growth mechanism involves an alternated bidimensional nucleation with a consecutive growth of a homochiral domain. The explanation for the oscillating crystallization is the following: the crystal growth leads under the condition of no stirring to an increasing diffusion layer at any crystallization face which finally increases the concentration of the enantiomer which is not incorporated in the crystal to an extend creating at the crystal surface a bidimensonal nucleation of the counter - enantiomer. An appropriate stirring rate led to crystals with a high enantiomeric excess. Dr. Michel Ollivon, University Paris-Sud, F gave a Communication on "Simultaneous Coupling of DSC and Time-Resolved Synchroton X-ray Diffraction for the Study of Colloides of Pharmaceutical Interest". Self-organization is one of the main features of long chain compounds such as lipids and surfactants from which most of the colloidal particles are constituted or stabilized. The interaction with and incorporation of water with these organic molecules is a decisive fact of the structure formation (lyotropism). The molecules bearing both long-chains and polar or ionised groups such as phospholipids or surfactants self pack in supramolecular multidimensional structures or aggregates like micelles, vesicles, emulsions. This class of polar molecules displays both lyotropic and thermotropoic behaviours (see phase diagrams e.g. DPPC-water). The diversity of the structures formed is imposed by the different possibilities of local curvature of the oil-water interface. These structures are stable or metastable with lifetimes spreading from less than one second to years. Research and characterization of these supramolecular structures are preferably made by a new technique, namely by time-resolved synchroton X-ray diffraction coupled with DSC. Several examples were outlined. G. Patrick Stahly, SSCI, West Lafayette, USA presented one example of a special crystallization pathway "Crystallization in Capillary Spaces: Preparation and Characterization of a Novel, High-Energy Polymorph of Nabumetone". The generation of seeds of high free energy polymorphs often requires special conditions. A possibility is the crystallization from solution in a limited volume and in a thin layer (capillary, plates). For Nabumetone a metastable form II was crystal-lized under such conditions which is converting into the known form I under mechanical stress. The method has a certain potential in the study of polymorphs. Professor Jean-Pierre Grolier, University Blaise-Pascal, Aubiere, F reported on the subject "The Combination of UV/VIS/IR Spectroscopy with Isothermal Fluxmetry and Isoperibolic Calorimetry to Perform and Monitor Polymerization in Organic Media". Anionic polymerization of lactams in non-polar solvents occurs through the "activated monomer mechanism" featuring a particular two-step propagation mechanism and leads to semicrystalline microscopic fine polyamid powders. Combining two different approaches, namely UV/VIS/IR spectroscopy and calorimetry allows to elucidate the kinetic pathway and to develop a monitoring of such a polymerisation. Two different calorimeters were used, the Calvet type C80 and the DRC reaction calorimeter. The in situ spectroscopic detection was possible through the use of optical fibers connected to spectrometers. The calorimeters were used as reactors, in which chain initiators and catalysts could be fed under controlled conditions. The calorimetric and spectroscopic response to a one-shot or to a continuous addition of active species could be analyzed in detail. Continuous addition of the catalyst led after an initial phase to a constant calorimetric signal because the reaction reached a steady state. The elucidation of the polymerization kinetics by the two approaches proofed to be rather successful. Dr. Anke Weidenkaff, University of Augsburg, Germany talked about "Microemulsions as Reactors for Nanoscopic Materials". Inorganic nanoparticles for catalytic applications are synthesized by co-precipitation processes in submicrometer-sized aqueous domains of reverse microemulsions. To tailor the particle structure, morphology, and surface area of metal oxides, studies on the influence of synthesis parameters such as composition of the microemulsion (reactants, surfactants, water-oil ratio) aging time, drying and calcination temperature of the precipitates have been outlined in detail. Professor Elena Boldyreva, Novosibirsk State University, Russia outlined the extraordinary interesting subject "Intermolecular Interactions in Solid Drugs - What Can High-pressure Techniques Contribute to their Study?". Intermolecular interactions play an important role in crystallization of drugs and in their properties such as stability, heat capacity, solubility, bioavailability, mechanical properties. Several examples were discussed in the study of crystals under high hydrostatic pressure with X-ray diffraction and vibrational spectroscopy. Special attentions were given to the role of hydrogen bond interactions with the anisotropy of structural distortion.
|
||||
| 4.3
Our
Sponsors.
TA
Instruments | ||||
| Company
Description
After 28 years as Du Pont Instruments, TA Instruments began independent operations in July 1990. Today, as a division of Waters Corporation, it is the world’s largest supplier of thermal analysis and rheology instruments. Its major thermal analysis products include the Q Series DSC Modules with Tzeroä technology (Q1000, Q100, Q10), Q Series TGA Modules (Q500, Q50), SDT Q600, Q800 DMA, TMA 2940, DEA 2970 and m -TA 2990. TA Instruments’ rheology products include the AR 2000 Advanced Rheometer, the AR 1000 Research Rheometer, the AR 500 Rheometer, and the QCR II Quality Control Rheometer. Headquartered in New Castle (DE. USA), TA Instruments is a worldwide company with subsidiaries throughout Europe, Japan and Australia, and factory trained agents / distributors in other countries. For more information contact TA Instruments Germany at the following address: Max
Planck
Strasse
11
| ||||
|
Differential Scanning Calorimetry (DSC) DSC is an analytical technique that measures heat flow as a function of sample temperature, and is the most commonly used thermal technique by the pharmaceutical industry and its suppliers. It is used in both product research and quality control for characterizing material properties, such as glass transition temperatures (lyophilization), melting temperatures, transition enthalpies, specific heat, polymorphism, purity, thermal and oxidative stability. DSC and other complementary thermal techniques (TGA, SDT, TMA, DMA, DEA, m -TA) are also used in the analysis of polymers associated with pharmaceutical products in encapsulation and packaging. TA rheometers measure flow and viscoelastic properties, which relate to correct formulation, processing, quality, and prediction of end-use properties. The identification and measurement of the glass transition temperature (Tg) is certainly one of the more important thermal measurements made in the pharmaceutical industry. Its accurate determination alone can lead to extensive cost savings in lyophilization (freeze drying) proceduresą. However, to accurately and precisely detect the glass transition temperature range, it is important to employ a DSC that offers a good and stable instrumental baseline, as discussed in the following technical paper by Dr. Bruce Cassel, a recognized authority on the subject. ą L.C Thomas, R.L.Blaine and G. Dallas, Today’s Chemist at Work, January 2002, pps 21-22.
| ||||
| New
DSC
Technology
in
the
Analysis
of
Physical
Aging
and
Fragility
of
Amorphous
Sucrose R. Bruce Cassel, TA Instruments, 109 Lukens Drive, New Castle, DE 19720, USA Amorphous materials undergo order of magnitude changes in viscoelastic properties when they are heated or cooled through the glass transition region (1). As an established materials characterization technique, DSC has long been used to determine a value for the glass transition temperature (Tg). However, unlike the melting point of a crystalline substance, which marks a well-defined change in the phase and is associated with a single temperature, the glass transition marks a temperature range over which there is a continuous change in the dynamic properties of the molecules. Moreover, the properties of the material below the glass transition region are dependent on the thermal history of the material as it was cooled from the liquid state. In particular, the rate of cooling from the melt, or the amount of annealing at temperatures in or near the glass transition region, effect the molar volume, enthalpy and viscoelastic properties of the material in the glassy state. Since the classic work of Tool (2) in quantifying glass transition behavior, there have been numerous efforts to model the glass transition process to rationalize the physical properties. Glass transition theoreticians aside, interest in this topic surfaces periodically in connection with "unexpected" changes in material properties when an amorphous material is stored above, within, or even below, the glass transition temperature. At temperatures below the glass transition a material may appear solid and in a stable state. In fact, if it is close to the glass transition, the material undergoes measurable changes in thermodynamic and viscoelastic properties that are referred to as physical aging. As a result of those changes the increased molecular mobility may allow crystallization or reaction to occur. While physical aging occurs to some degree with all amorphous materials, the degree to which it occurs, and thus how far below Tg a material must be stored to be stable, varies by an order of magnitude depending on the material (3). Predicting this for a particular material, quantifying it, and looking for ways to modify it have become of interest in connection with the stability of amorphous pharmaceutical formulations. The glass transition temperature is a somewhat arbitrary value assigned to a measurable point within the glass transition region as defined by a specific procedure. In general, the value of Tg depends on the analytical technique, for example, calorimetry, volumetric analysis, or rheology. It also depends on the time scale of the measurement, e.g., the DSC heating rate, or MDSC® or DMA frequency, and in addition on the previous thermal (and mechanical) history of the sample. One definition of Tg that has particular value for understanding physical aging is the fictive temperature (Tf), which has been defined as the extrapolated intersection of the pre-transition and post-transition DSC baselines in enthalpy units (1). To obtain the enthalpy functions one integrates the DSC heat flow curves. There is also an equivalent graphical method of obtaining Tf directly from the DSC trace (4). Figure 1 shows the Tf calculation on a DSC specific heat capacity trace and on its integral. The unique property of the fictive temperature is that it is independent of the DSC heating rate used to measure it. Hence, it gives a value for the glass transition that depends only on the previous cooling rate through the transition region, which determines the enthalpy state of the material below Tg. The term used to describe the sensitivity of a material to physical aging is "fragility". Fragility for an amorphous material can be defined in terms of changes measured by a rheometer, dilatometer or DSC. As measured by DSC, the enthalpic fragility parameter (3) is defined as: mDh = - (d log bc)/(d(Tf,ref /Tf)), (eq. 1) where | ||||
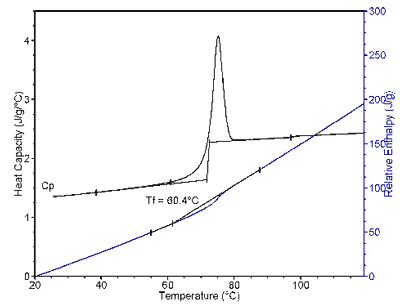 | Figure
1.
Example of fictive temperature calculation using heat capacity or relative enthalpy data of sucrose (19.9 mg sample heated at 15 K/min after cooling at 0.1 K/min). | |||
|
Preparation of starting material: The 19.9 mg crystalline sucrose sample was encapsulated in a standard crimped DSC pan and heated to 210°C in nitrogen to produce the melt. The sample then was removed and placed on a conductive surface at room temperature. This rapid cool-down procedure prevented crystallization of the sample, thus "trapping" it in the amorphous state. The data below was obtained from cycling this sample specimen through the Tg region. Specific heat capacity (Cp) data show no evidence of appreciable change in the amorphous content due to crystallization. Instrumentation: A Q 1000TM DSC with Advanced TzeroTM Technology was used for the analysis. This new DSC and sensor technology has been described in a number of publications (5), and its technical capabilities are particularly useful for this method. After calibration, the temperature displayed and analyzed is essentially the temperature of the DSC pan holding the sample. Hence, for a sample that is well coupled to the pan, all thermal lag has been compensated in the abscissa data. This allows data to be compared at different heating and cooling rates with confidence that the DSC is calibrated for all these conditions (6). A critical feature for this analysis is the absolute stability and linearity of the baseline. Because the fictive temperature determination requires an extrapolation across the glass transition interval, it is essential that the baseline be devoid of curvature. The Q Series DSC cell and sensor design provides this capability. Also necessary are rapid equilibration and fast cooling rates, and both of these performance features are substantially improved in the Q Series DSC analyzer systems. | ||||
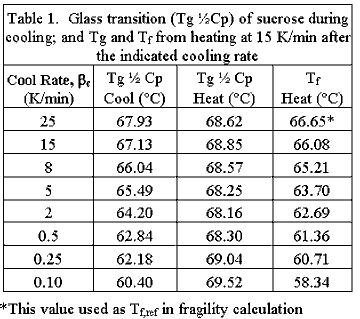
|
Another advantage is that with the removal of the instrumental baseline from the DSC output, the signal can be fully normalized into units of specific heat capacity, which is the fundamental thermodynamic property of the material being measured. The measurements of the DSC analysis on sucrose can be seen in Figure 2 and the results are presented in Table 1. The collection of data for Table 1 was obtained in a single, overnight, multiple-step program that alternately conditioned the sample through the glass transition region at successive cooling rates and alternately heated it at a fixed rate of 15 K/min. | |||
|
Figure
1
shows
an
example
of
a
calculation
performed
on
one
of
the
heating
steps.
The
glass
transition
was
measured
at
the
indicated
cooling
rates
using
the
commonly
used
˝
Cp
glass
transition
assignment
protocol
(7).
The
Tg
was
measured
in
the
subsequent
heating
step
using
both
the
fictive
temperature
method
used
for
the
fragility
calculation
and
the
˝
Cp
method.
Notice
that
the
Tg
is
depressed
both
when
measured
in
cooling
and
under
the
fictive
protocol
when
subsequently
heated.
This
downward
shift
is
consistent
with
the
changes
in
other
physical
properties,
which
accompany
physical
aging,
namely
increased
mobility
at
lower
temperatures.
The
slow
change
in
mobility,
which
attends
physical
aging,
leads
to
stability
problems
for
long-term
storage
of
amorphous
formulations. | ||||
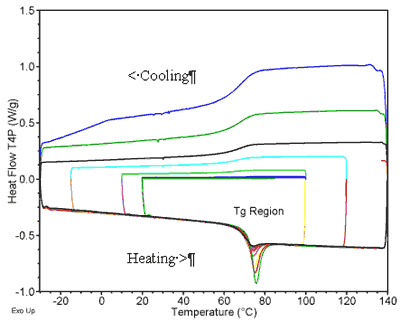 |
Figure
2.
Raw DSC data for determining sucrose fragility, obtained in a multiple-step method that alternates cooling at successive rates and heating at a fixed rate of 15 K/min. | |||
|
In
contrast
to
the
fictive
and
cooling
data,
the
standard
Tg
method
does
not
show
this
trend
at
a
heating
rate
of
15°
K/min
because
of
kinetic
delay.
See
Figure
3.
Heating
at
a
very
slow
rate
gives
Tg
data
approaching
that
of
Tf.
One
of
the
advantages
of
the
˝
Cp
method
for
determining
Tg
is
that
it
is
less
sensitive
to
physical
aging
changes
and
therefore
more
likely
to
give
a
material-dependent
measure
of
the
midpoint
of
the
glass
transition
region. | ||||
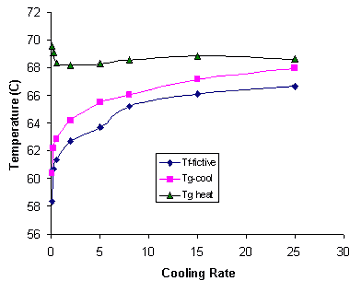 |
Figure 3.
Glass transition of sucrose from cooling at several rates, and Tg and Tf from subsequent heating at 15 K/min. | |||
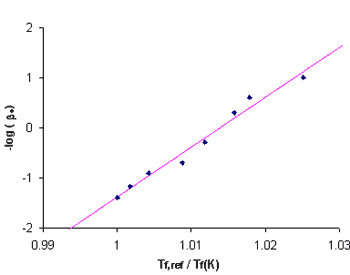
|
Figure 4.
Fragility plot of Sucrose using the above Tf data and a Tf,ref of 66.65°C taken from the 25 K/min-cooled sucrose. | |||
|
Using
Equation
1
and
the
fictive
data
from
Table
1,
the
fragility
parameter,
mDh,
for
sucrose
was
calculated
as
100
(+/-2)
using
the
least
squares
fit
method.
From
equation
1,
we
can
predict
that
physical
aging
for
three
weeks
(a
cooling
rate
through
Tg
of
0.001
K/min)
would
produce
a
Tf
of
53şC.
Storage
above
this
temperature
would
be
expected
to
provide
the
mobility
for
crystallization
or
reaction.
Robertson
noted
that
the
fragility
could
have
been
calculated
directly
from
the
cooling
Tg
data
(shown
in
the
first
column
of
Table
1)
but
for
problems
of
sensitivity
and
temperature
calibration
in
cooling,
where
correction
for
thermal
lag
is
normally
different
at
each
rate.
All
of
these
issues
in
analyzing
the
cooling
data
have
been
resolved
with
the
technology
in
the
Q1000
DSC. | ||||
| SUMMARY
The
above-described
method
has
been
used
to
calculate
the
fragility
of
sucrose.
This
approach
could
be
extended
(by
choice
of
heating
and
cooling
ranges)
to
allow
determination
of
the
fragility
parameter
for
other
glass
formers,
such
as
excipients,
amorphous
drug
substances
or
amorphous
formulations.
The
Q1000
DSC
with
Advanced
TzeroTM
Technology
has
considerable
advantage
for
this
analysis
because
of
its
improved
temperature
control,
accuracy,
and
baseline
stability. | ||||
|
REFERENCES
| ||||
| ||||